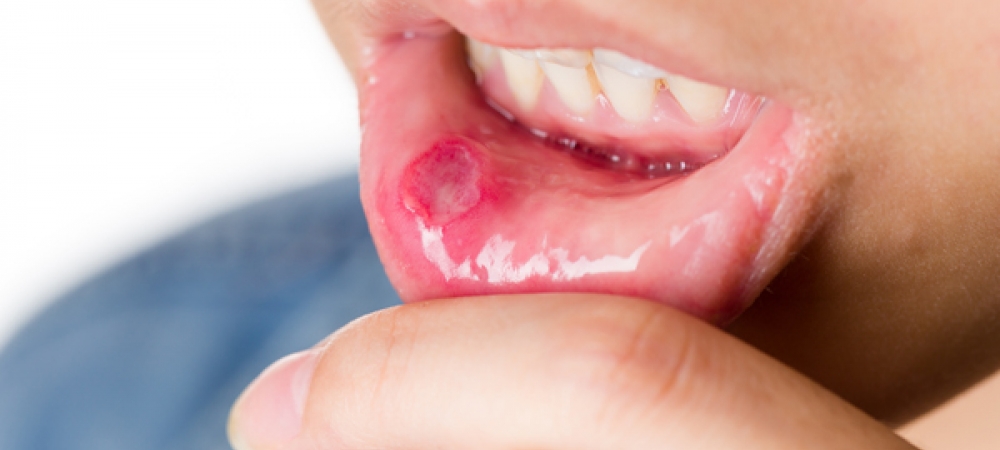
A doença de Behçet (CID: M35.2) é uma doença inflamatória rara que pode afetar várias partes do corpo ao mesmo tempo. Ela se caracteriza pela presença de úlceras aftosas na boca e na região genital, além de inflamações nos vasos sanguíneos, que podem ser tanto artérias quanto veias, de diferentes tamanhos.
Por atingir diferentes sistemas do organismo, a doença de Behçet é considerada uma doença multissistêmica. Seu diagnóstico costuma ser clínico, a partir da combinação dos sintomas, e o acompanhamento médico é essencial para o controle da condição.
Causas da doença
A causa da doença ainda é desconhecida. No entanto, especialistas acreditam que a doença começa quando o sistema imunológico reage de forma exagerada, possivelmente após um gatilho ambiental, como infecções por vírus ou bactérias. Essa resposta autoimune acontece em pessoas que já têm uma predisposição genética.
Entre os fatores genéticos, o mais associado ao risco de desenvolver a doença de Behçet é o HLA-B51, que está presente em cerca de 20% dos casos.
Sintomas: como a doença se manifesta?
Os sintomas da doença geralmente começam entre o final da puberdade e os 40 anos de idade. Pode afetar homens e mulheres em proporções semelhantes, mas costuma evoluir de forma mais grave nos homens, com manifestações mais intensas em diferentes partes do corpo.
Manifestações cutâneas e mucosas
Um dos sinais mais característicos são as úlceras na boca e na região genital. Essas lesões são recorrentes e costumam surgir em surtos que duram de 2 a 6 semanas.
-
Aftas orais: geralmente maiores que 0,5 cm, com bordas avermelhadas e fundo escavado, podendo afetar toda a orofaringe.
-
Úlceras genitais: parecidas com as orais, são mais comuns na bolsa escrotal (em homens) e nos pequenos lábios e parede vaginal (em mulheres).
-
Lesões de pele: os tipos mais frequentes são o eritema nodoso e a pseudofoliculite.
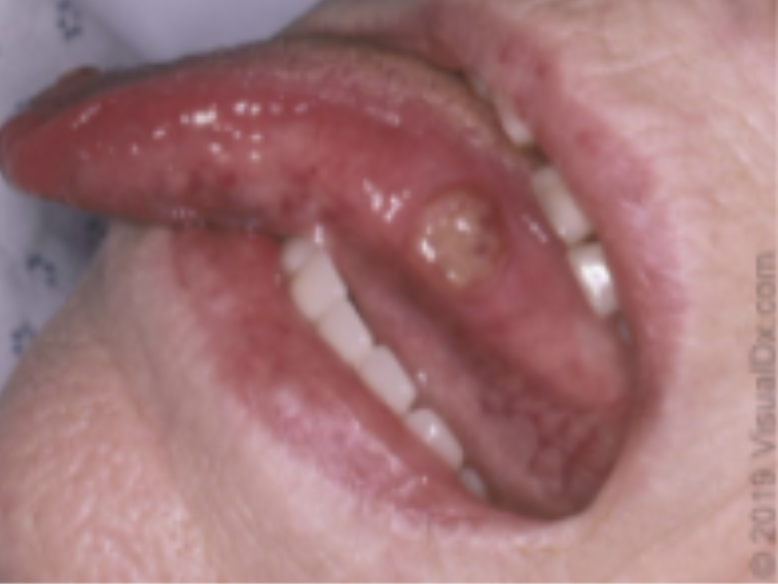
Úlcera oral em língua (Fonte: Uptodate 2021)
Manifestações oculares
Entre 50% e 70% das pessoas com doença de Behçet apresentam alterações nos olhos, principalmente nos primeiros anos da doença.
-
O tipo mais comum é a uveíte bilateral, que pode ser anterior, posterior ou do tipo panuveíte.
-
Também pode ocorrer vasculite na retina, o que exige acompanhamento oftalmológico cuidadoso.
Manifestações articulares
Cerca de 40% a 80% dos pacientes apresentam sintomas nas articulações.
-
O quadro costuma afetar poucos locais (mono ou oligoarticular), de forma assimétrica, principalmente joelhos e tornozelos.
-
Dores musculares (mialgia) também são comuns.
Manifestações neurológicas
As alterações neurológicas podem aparecer no início da doença, sendo a primeira manifestação em até 20% dos casos. Elas incluem:
-
Meningoencefalite asséptica
-
Vasculite cerebral
-
Acidente vascular encefálico (AVE)
-
Trombose venosa central
-
Aneurismas
Manifestações vasculares
A doença de Behçet também pode causar inflamações e tromboses em vasos sanguíneos importantes, como:
-
Veias de grande calibre, como as hepáticas, cava e veias profundas dos membros
-
Artérias, como carótidas, pulmonares, aorta, ilíaca e femoral — com possibilidade de formação de aneurismas.
Diagnóstico
O diagnóstico da doença de Behçet é feito exclusivamente com base na avaliação clínica. Ou seja, não existem exames específicos que confirmem a doença. O médico chega ao diagnóstico observando os sintomas e o histórico do paciente.
Para fins de estudo e padronização, utiliza-se o Critério Internacional de Classificação da Doença de Behçet, que atribui uma pontuação a cada manifestação da doença:
Critério Internacional de Classificação da Doença de Behçet
| Sinal ou sintoma | Pontos |
|---|---|
| Lesões oculares | 2 |
| Lesões genitais | 2 |
| Aftas orais | 2 |
| Lesões cutâneas | 1 |
| Manifestações neurológicas | 1 |
| Manifestações vasculares | 1 |
| Teste de patergia (opcional) | 1 |
O diagnóstico é estabelecido quando a soma total for igual ou superior a 4 pontos.
Diagnósticos diferenciais
Outras condições que podem se parecer com a doença de Behçet incluem:
-
Estomatite aftosa recorrente
-
Outras vasculites
-
Síndrome do anticorpo antifosfolípide
-
Doença de Crohn
O que é o teste de patergia?
O teste de patergia consiste em observar a pele após uma pequena lesão provocada por uma agulha fina. Se surgir uma reação inflamatória local, como uma pústula ou pápula avermelhada em até 48 horas, o resultado é considerado positivo.
Essa reação é mais comum em pessoas de regiões onde a doença é endêmica, como nos países asiáticos da Rota da Seda, onde a taxa de positividade pode chegar a 50%–75%. No entanto, o teste não é obrigatório para o diagnóstico.
Diagnósticos diferenciais importantes são: estomatite aftosa recorrente, outras vasculites, síndrome do anticorpo antifosfolípide, doença de Chron.
Tratamento
A doença de Behçet é uma doença autoimune crônica, o que significa que não tem cura, mas pode ser controlada com o tratamento adequado.
A escolha do tratamento depende dos sintomas apresentados e dos órgãos afetados. Os medicamentos mais usados incluem:
-
Colchicina
-
Azatioprina
-
Ciclosporina
-
Ciclofosfamida
-
Imunobiológicos, como os inibidores do fator de necrose tumoral (anti-TNF)
O quadro clínico costuma evoluir com fases de crise (exacerbação) e melhora (remissão). Com o tempo, geralmente após cerca de 10 anos de sintomas ou após os 40 anos de idade, a atividade da doença tende a diminuir gradualmente.
Para mais informações sobre o tratamento da doença de Behçet, agende sua consulta!
