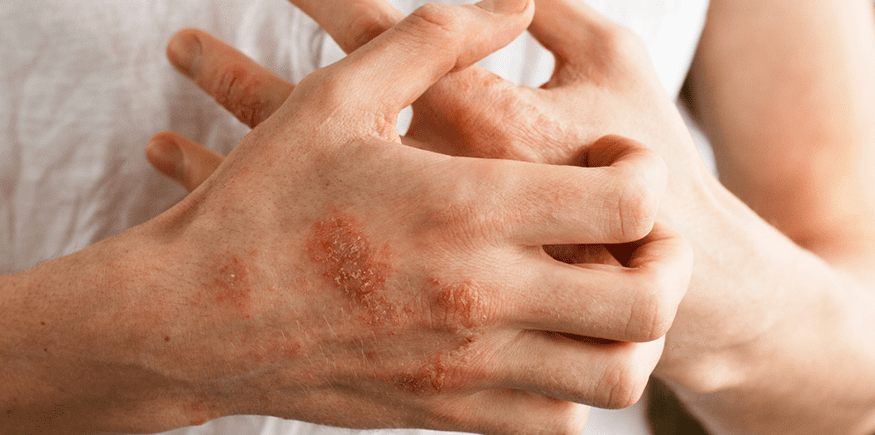
A esclerodermia é uma doença rara que afeta o tecido conjuntivo da pele, músculos, articulações e órgãos internos.
Ela causa inflamação, endurecimento e afinamento dos tecidos, além de alterações nos vasos sanguíneos, que podem levar à redução da circulação. A doença é mais comum em mulheres e geralmente começa entre os 45 e 64 anos.
Causas da Esclerodermia
A origem da esclerodermia é complexa e envolve diferentes fatores. O sistema imunológico, o comprometimento dos vasos sanguíneos e a produção excessiva de colágeno desempenham um papel importante no desenvolvimento da doença.
Cerca de 95% dos pacientes com esclerodermia apresentam autoanticorpos no sangue, que atacam estruturas do próprio organismo. Os principais são:
- anti-Scl 70 (antitopoisomerase I)
- anticentrômero
- anti-RNA polimerase III
- antifibrilarina
- anti-PM-Scl.
Sintomas e manifestações clínicas
A esclerodermia pode se manifestar de diferentes formas. É fundamental distinguir entre a esclerodermia localizada, que afeta principalmente a pele, e a esclerose sistêmica, que pode comprometer órgãos internos além da pele.
Esclerodermia localizada
Também conhecida como morfeia, é uma afecção que acomete a pele e os tecidos adjacentes, caracterizada principalmente pelo espessamento cutâneo.
Não há acometimento de órgãos internos e normalmente tem curso benigno e autolimitado. Pode se apresentar como:
- Morfeia em placas: lesões circunscritas de coloração eritematosa ou violácea.
- Morfeia linear: induração linear acometendo tronco, membros ou face (golpe de sabre). Mais comum em crianças.
- Morfeia generalizada: placas confluentes extensas.
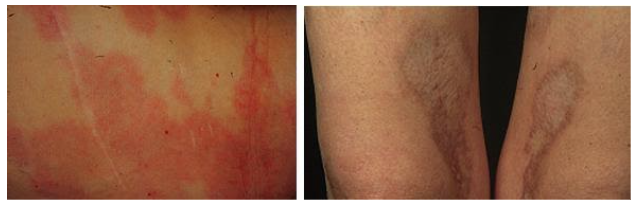
Esclerose sistêmica
O diagnóstico deve ser considerado principalmente em pacientes do sexo feminino, com fenômeno de Raynaud, espessamento cutâneo, fadiga, artralgias, dispneia (falta de ar) ou disfagia (dificuldade para engolir).
Existem duas formas clássicas de apresentação da esclerose sistêmica:
- ES cutânea limitada: espessamento cutâneo restrito às extremidades dos membros e face. Tipicamente associado ao anticorpo anticentrômero. Evoluiu com hipertensão arterial pulmonar em fases mais tardias.
- ES cutânea difusa: espessamento cutâneo precoce e se estende à região proximal dos membros e tronco. Fibrose pulmonar e crise renal esclerodérmica são mais frequentes e os anticorpos encontrados são o anti-Scl70 e o anti-RNA polimerase III.
O que é Fenômeno de Raynaud?
O fenômeno de Raynaud (FRy) é caracterizado por um vasoespasmo reversível das extremidades, por exposição ao frio ou ao estresse emocional. Manifesta-se clinicamente por dedos frios associados a mudanças de cor bem demarcadas da pele: primeiro ocorre uma vasoconstrição (palidez) seguindo pela cianose (arroxeado) e, por fim, reperfusão (rubor).
Geralmente começa em um único dedo e, em seguida, se espalha para outros dedos simetricamente em ambas as mãos. O indicador, o dedo médio e o anular são os dedos mais frequentemente envolvidos, enquanto o polegar é costuma poupado.
O FRy é reversível com o reaquecimento ou com a redução do estresse.
Pode ser classificado em:
- FRy primário: presente em 3 a 5% da população, principalmente em mulheres jovens e com história familiar positiva. Não há evidência de causa secundária, doença vascular periférica, isquemia digital ou anormalidade na capilaroscopia. Sem dano estrutural na microcirculação.
- FRy secundário: presença de alguma comorbidade ou causa associada à ocorrência do fenômeno, como:
- Doenças autoimunes: esclerose sistêmica, lúpus eritematoso sistêmico, síndrome de Sjogren, hipotireoidismo
- Doenças hematológicas: síndromes paraneoplásicas, crioglobulinemia, doença das criaglutinindas
- Causas vasculares: embolia, vasculite, aterosclerose, tromboangeíte obliterante
- Medicamentoso: cocaína, nicotina, medicações para enxaqueca, clonidina, betabloqueadores
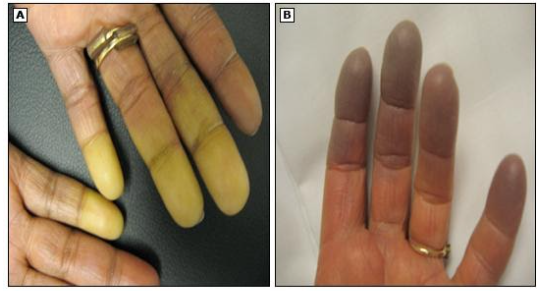
B- Cianose digital da ponta dos dedos resultante da vasoconstrição.
Fonte: Uptodate 2021
Diagnóstico
A esclerodermia é uma doença rara e um dos maiores desafios na prática dos reumatologistas. Seu diagnóstico é feito através da história clínica, achados ao exame físico, métodos de imagem e exames laboratoriais.
A presença isolada e inicial do FRy, associado à presença de autoanticorpos específicos (anti-Scl70 ou anticentrômero ou anti-RNA polimerase III) e/ou capilaroscopia anormal, configura a fase precoce ou muito precoce da doença.
Além do acometimento cutâneo característico, é possível haver: alterações pulmonares, no trato gastrointestinal (principalmente esôfago) nos rins e no aparelho musculoesquelético.
Tratamento
A esclerose sistêmica é uma doença crônica autoimune e que não apresenta cura. Seu tratamento, em geral, é determinado pela extensão e gravidade das manifestações clínicas.
O avanço nas estratégias de diagnóstico tem proporcionado uma identificação mais precoce dos casos e, com isso, início de terapias imunossupressores antes da progressão do quadro fibrótico. As medicações utilizadas dependem do tipo de manifestação clínica do paciente.
Imunossupressores para acometimento cutâneo e pulmonar, inibidores de bomba de prótons e procinéticos para sintomas gastrointestinais, bloqueadores de canal de cálcio para FRy e uso de inibidores de fosfodiesterase 5 e/ou antagonistas dos receptores de endotelina para hipertensão arterial pulmonar.
CID: M 34; M 34.1; M34.8; M 34.9; M 34.0
